R语言进行TCGA配对样本差异基因分析_r语言差异分析-程序员宅基地
技术标签: python 数据分析 机器学习 人工智能 数据可视化
之前的一个推文是从UCSC XENA获取TCGA的表达和表型数据,然后利用代码对表达数据进行了ID注释,以及mRNA、lncRNA和miRNA的区分筛选,最后将患者ID和临床信息进行配比,用于后续分析。详细内容见推文:《利用R代码从UCSC XENA下载mRNA, lncRNA, miRNA表达数据并匹配临床信息》。
本期我将继续上次的内容,从TCGA 546个头颈癌数据集(Tumor = 502,Normal = 44)中,提取出43对癌和癌旁样本,并做配对差异分析, 然后绘制某个基因的配对箱式图。实际上TCGA好多癌症比如头颈癌、肝癌等,都有癌和癌旁的数据,且癌和癌旁都是一一配对的关系。所以在分析癌vs.癌旁的过程中,可以选择普通的差异分析,例如头颈癌的502个癌vs.44癌旁;另外一种思路是从中挑选出配对的癌vs.癌旁进行配对差异分析,例如头颈癌的43个癌vs.43个癌旁。实际上,同一个数据的非配对分析和配对分析差异还是很大的,详见我之前写过的一个帖子《差异分析|DESeq2完成配对样本的差异分析》。
关于DEseq2配对差异分析,很少有帖子涉及 (生信宝典注:不是很少有帖子涉及,而是看有没有发现,配对的个体信息可以视作一个批次因素,在DESeq2差异基因分析和批次效应移除, 高通量数据中批次效应的鉴定和处理 - 系列总结和更新, 典型医学设计实验GEO数据分析 (step-by-step) - Limma差异分析、火山图、功能富集中都有类似的处理方式)。
总结来说,拿到配对设计的数据,如果不进行配对分析而用常规的差异分析,这样的结果可能会大不相同。因此,在分析数据的时候,一定要明白实验设计。
下面代码展示了如何从546个样本中挑选出一一配对的癌和癌旁数据,并进行DEseq2配对差异分析。
remove(list = ls())
##加载包
suppressMessages(library(DESeq2))
suppressMessages(library(dplyr))
library(ggplot2)
library(ggpubr)
library(ggthemes)1. 加载数据
首先,读入上期推文中处理好的表达矩阵(仅含mRNA)和表型数据。
# 1.1 表达矩阵
expr_data <- read.csv("./Rawdata/TCGA_mRNA_count_log2.csv",header = T,row.names = 1,check.names=F)
expr_data[1:3,1:4]
## TCGA.BB.4224.01 TCGA.H7.7774.01 TCGA.CV.6943.01 TCGA.CN.5374.01
## A4GNT 2.584963 0.00000 1.00000 2.321928
## AAAS 10.952741 10.43463 11.27671 10.750707
## AACS 11.098032 13.62822 12.52943 11.171802
# 1.2 表型数据
phenotype <- read.csv("./Rawdata/TCGA_HNSC_phenotype.csv",header = T,row.names = 1,check.names=F, stringsAsFactors = T)
phenotype$subject <- as.factor(substring(rownames(phenotype),1,12))
phenotype[1:3,c("group","subject")]
## group subject
## TCGA.BB.4224.01 Tumor TCGA.BB.4224
## TCGA.H7.7774.01 Tumor TCGA.H7.7774
## TCGA.CV.6943.01 Tumor TCGA.CV.6943
# 检查一下表达数据和表型数据是否是一一对应
table(colnames(expr_data) == rownames(phenotype)) #T
##
## TRUE
## 546表达数据和表型数据如果不是一一对应的话,运行下面这行代码:
# 利用match函数进行匹配
phenotype <- phenotype[match(colnames(expr_data),rownames(phenotype)),]
identical(colnames(expr_data),rownames(phenotype))
## [1] TRUE2. 筛选出配对的样本
2.1 将546个样本分为两部分,Tumor组和Normal组
# Tumor组
Tumor_group <- phenotype[which(phenotype$group == "Tumor"),] #TCGA的Tumor样本,502个
table(Tumor_group$group) #502个癌
##
## Nontumor Tumor
## 0 502
# Normal组
Normal_group <- phenotype[which(phenotype$group == "Nontumor"),] #TCGA的control样本,44
table(Normal_group$group) #44个癌旁
##
## Nontumor Tumor
## 44 0
# 找到癌和癌旁能够进行一一配对的样本的信息
Normal_group <- Normal_group[Normal_group$subject %in% Tumor_group$subject,]
#仅保留配对的对照组
table(Normal_group$group) # 43个存在配对的癌旁
##
## Nontumor Tumor
## 43 0上面这行代码就获取了43个配对的癌旁样本信息,根据这个信息可以获得所有配对的癌+癌旁,代码如下:
# 获取表达矩阵
paired_expr_data <- expr_data[,substring(colnames(expr_data),1,12) %in%
substring(rownames(Normal_group),1,12)]
# 实际上,这里的substring(rownames(Normal_group),1,12)和Normal_group$ID是一样的
paired_phe <- phenotype[match(colnames(paired_expr_data),row.names(phenotype)),]
paired_phe <- droplevels(paired_phe)
dim(paired_expr_data) # 获得了86个样本表达信息,其中43个癌,43个癌旁,是一一配对的关系
## [1] 18192 86
write.csv(paired_expr_data,"./Output/TCGA_HNSC_mRNA_count_paired_43vs43.csv")然后,进行配对的DEseq2差异分析。这里只保留了至少在75%的样本中都有表达的基因。
3. DEseq2配对差异分析
3.1 配对的表达矩阵
# UCSC Xena下载的数据是经过log2(count+1)处理的
data = (2^paired_expr_data-1) %>% apply(2, as.integer)# 还原log2的处理
row.names(data) = row.names(paired_expr_data)
# 保留至少在75%的样本中都有表达的基因
keep_data <- rowSums(data> 0) >= floor(0.75*ncol(data))
table(keep_data)
## keep_data
## FALSE TRUE
## 2599 15593
data <- data[keep_data,]
data[1:4,1:4]
## TCGA.CV.6943.01 TCGA.CV.6959.01 TCGA.CV.7438.11 TCGA.CV.7242.11
## AAAS 2479 3304 1900 2190
## AACS 5911 3881 4759 3070
## AADAC 7 5 449 2618
## AADAT 140 77 160 127
dim(data)
## [1] 15593 863.2 对应的表型数据和配对信息
table(paired_phe$subject,paired_phe$group)
##
## Nontumor Tumor
## TCGA.CV.6933 1 1
## TCGA.CV.6934 1 1
## TCGA.CV.6935 1 1
## TCGA.CV.6936 1 1
## TCGA.CV.6938 1 1
## TCGA.CV.6939 1 1
## TCGA.CV.6943 1 1
## TCGA.CV.6955 1 1
## TCGA.CV.6956 1 1
## TCGA.CV.6959 1 1
## TCGA.CV.6960 1 1
## TCGA.CV.6961 1 1
## TCGA.CV.6962 1 1
## TCGA.CV.7091 1 1
## TCGA.CV.7097 1 1
## TCGA.CV.7101 1 1
## TCGA.CV.7103 1 1
## TCGA.CV.7177 1 1
## TCGA.CV.7178 1 1
## TCGA.CV.7183 1 1
## TCGA.CV.7235 1 1
## TCGA.CV.7238 1 1
## TCGA.CV.7242 1 1
## TCGA.CV.7245 1 1
## TCGA.CV.7250 1 1
## TCGA.CV.7252 1 1
## TCGA.CV.7255 1 1
## TCGA.CV.7261 1 1
## TCGA.CV.7406 1 1
## TCGA.CV.7416 1 1
## TCGA.CV.7423 1 1
## TCGA.CV.7424 1 1
## TCGA.CV.7425 1 1
## TCGA.CV.7432 1 1
## TCGA.CV.7434 1 1
## TCGA.CV.7437 1 1
## TCGA.CV.7438 1 1
## TCGA.CV.7440 1 1
## TCGA.H7.A6C4 1 1
## TCGA.HD.8635 1 1
## TCGA.HD.A6HZ 1 1
## TCGA.HD.A6I0 1 1
## TCGA.WA.A7GZ 1 13.3 配对差异分析
# DEseq2的配对差异分析跑的有点慢,43对我的电脑需要约10分钟
# 所以,这里我为了加快运行速度,选了5对5.
data = data [,order(paired_phe$subject)]
data = as.matrix(data[,1:10])
data[1:3,1:4]
## TCGA.CV.6933.01 TCGA.CV.6933.11 TCGA.CV.6934.01 TCGA.CV.6934.11
## AAAS 2993 1328 2816 1977
## AACS 5124 4997 3522 4108
## AADAC 98 1 16 63
coldata_paired <- paired_phe[colnames(data),]
coldata_paired[,c('subject','group')]
## subject group
## TCGA.CV.6933.01 TCGA.CV.6933 Tumor
## TCGA.CV.6933.11 TCGA.CV.6933 Nontumor
## TCGA.CV.6934.01 TCGA.CV.6934 Tumor
## TCGA.CV.6934.11 TCGA.CV.6934 Nontumor
## TCGA.CV.6935.01 TCGA.CV.6935 Tumor
## TCGA.CV.6935.11 TCGA.CV.6935 Nontumor
## TCGA.CV.6936.01 TCGA.CV.6936 Tumor
## TCGA.CV.6936.11 TCGA.CV.6936 Nontumor
## TCGA.CV.6938.01 TCGA.CV.6938 Tumor
## TCGA.CV.6938.11 TCGA.CV.6938 Nontumor
# 3.4.1 DEseq2的配对差异分析
dds_paired <- DESeqDataSetFromMatrix(countData = data,
colData = coldata_paired,
design = ~ subject + group)
## factor levels were dropped which had no samples
dds_paired$group <- relevel(dds_paired$group, ref = "Nontumor") # 指定哪一组作为对照
dds_paired <- DESeq(dds_paired)
## estimating size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
nrDEG_paired <- as.data.frame(results(dds_paired))
nrDEG_paired = nrDEG_paired[order(nrDEG_paired$log2FoldChange),] # 按照logFC排序
nrDEG_paired[1:3,1:6]
## baseMean log2FoldChange lfcSE stat pvalue padj
## PRH2 544288.00 -15.63870 2.509593 -6.231567 4.617911e-10 7.423411e-08
## STATH 1385580.80 -15.33676 1.292428 -11.866626 1.764414e-32 9.170836e-29
## MUC21 26826.61 -12.83806 1.239818 -10.354790 3.980606e-25 7.758699e-22
write.csv(nrDEG_paired,file = "./Output/TCGA_HNSCpaired_DESeq2_nrDEG.csv")
# 定义差异基因,用于后续的分析,例如火山图,GO和KEGG等
logFC_cutoff <- 2
nrDEG_paired$change <- as.factor(ifelse(nrDEG_paired$padj < 0.05 & abs(nrDEG_paired$log2FoldChange) > logFC_cutoff,
ifelse(nrDEG_paired$log2FoldChange > logFC_cutoff ,'UP','DOWN'),'NOT'))
table(nrDEG_paired$change)
##
## DOWN NOT UP
## 753 14452 388
# 绘制火山图
Volcano_data <- na.omit(nrDEG_paired)
Volcano_data$logP <- -log10(Volcano_data$padj)
Volcano_paired <- ggscatter(Volcano_data, x = "log2FoldChange", y = "logP",
color = "change",
palette = c("#2f5688", "#BBBBBB", "#CC0000"),
size = 1,
font.label = 10,
repel = T,
xlab = "log2 FoldChange",
ylab = "-log10 (pvalue)",
title="Volcano Plot") +
theme_base() +
geom_hline(yintercept = 1.30, linetype="dashed") +
geom_vline(xintercept = c(-logFC_cutoff,logFC_cutoff), linetype="dashed")+
theme(legend.position = "right",plot.title = element_text(size = 14,color="black",hjust = 0.5))
Volcano_paired
ggsave("./Output/Volcano_paired.PDF",Volcano_paired,width=6,height=5)上述就是配对DEseq2差异分析的一个结果。接下来,我们看一下如果使用普通DEseq2差异分析的结果,将会有哪些区别?
# 3.5 DEseq2的普通差异分析
dds_2 <- DESeqDataSetFromMatrix(countData = data,
colData = coldata_paired,
design = ~ group)
dds_2$condition <- relevel(dds_2$group, ref = "Nontumor") # 指定哪一组作为对照
dds_2 <- DESeq(dds_2)
## estimating size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
nrDEG_2 <- as.data.frame(results(dds_2))
nrDEG_2 = nrDEG_2[order(nrDEG_2$log2FoldChange),] # 按照logFC排序
nrDEG_2[1:3,1:4]
## baseMean log2FoldChange lfcSE stat
## PRH2 544288.0 -20.27705 2.722897 -7.446865
## STATH 1385580.8 -18.75144 2.069789 -9.059590
## PRH1 104842.6 -15.65276 1.820363 -8.598703
write.csv(nrDEG_2,file = "./Output/TCGA_HNSC_DESeq2_nrDEG.csv")
# 这里我还提取了标准化后的表达矩阵,可以用于后续的热图箱式图绘制等等,当然也可以使用FPKM或TPM代替
rld_paired <- vst(dds_paired)
Nor_expr_data_paired <- assay(rld_paired)
write.csv(Nor_expr_data_paired,file = "./Output/TCGA_HNSCpaired_Norexpr_data_paired.csv")
# 3.5 定义差异基因,用于后续的分析,例如GO和KEGG等
logFC_cutoff <- 2
nrDEG_2$change <- as.factor(ifelse(nrDEG_2$padj < 0.05 & abs(nrDEG_2$log2FoldChange) > logFC_cutoff,
ifelse(nrDEG_2$log2FoldChange > logFC_cutoff ,'UP','DOWN'),'NOT'))
table(nrDEG_2$change)
##
## DOWN NOT UP
## 626 14093 375
# 3.6 绘制火山图
Volcano_data_2 = na.omit(nrDEG_2)
Volcano_data_2$logP <- -log10(Volcano_data_2$padj)
Volcano_2 <- ggscatter(Volcano_data_2, x = "log2FoldChange", y = "logP",
color = "change",
palette = c("#2f5688", "#BBBBBB", "#CC0000"),
size = 1,
font.label = 10,
repel = T,
xlab = "log2 FoldChange",
ylab = "-log10 (pvalue)",
title="Volcano Plot") +
theme_base() +
geom_hline(yintercept = 1.30, linetype="dashed") +
geom_vline(xintercept = c(-logFC_cutoff,logFC_cutoff), linetype="dashed")+
theme(legend.position = "right",plot.title = element_text(size = 14,color="black",hjust = 0.5))
Volcano_2

ggsave("./Output/Volcano_2.PDF",Volcano_2 ,width=6,height=5)可以看到配对的DEseq2差异分析有753基因下调,388基因上调;而常规的差异分析有626个基因下调,375基因上调,这个结果看似差不多 (生信宝典注:这里应该做个Venn图比较下的,除了数量,具体基因是否一致。可参考高通量数据中批次效应的鉴定和处理 - 系列总结和更新)。
实际上,笔者曾处理过一个项目,用配对和不用配对,得到的结果相差甚远,主要还是体现在火山图、KEGG和GO分析等需要输入差异基因的分析中。但好像做GSEA这样纳入全部基因的分析,并不会受到太大的影响,这点有机会再探讨。
生信宝典注 (具体见一文掌握GSEA,超详细教程!):
-
如果用log2FC做GSEA结果是肯定会有差别的,影响多大不好说。
-
如果用标准化后的矩阵做GSEA结果没差别,因为配对分析没有影响到表达标准化,而只是在差异分析步骤起作用。
绘制单基因配对t检验箱式图
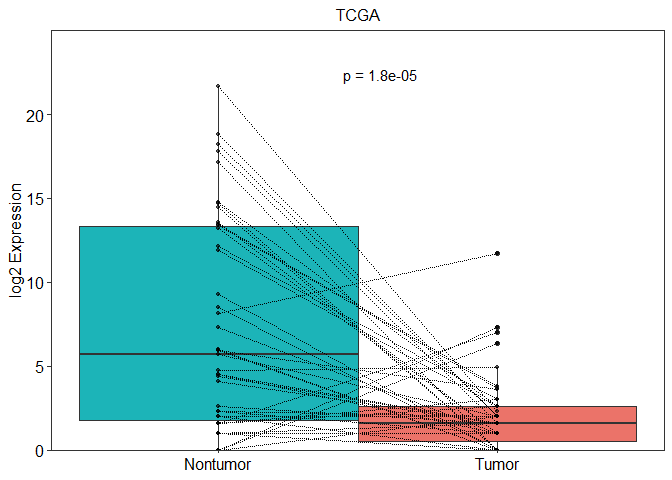
4.1 提取PRH2基因的表达量
## 4. PRH2表达量的配对箱式图绘制
PRH2_expression <- t(paired_expr_data["PRH2",] ) %>% data.frame(PRH2 = .)
PRH2_expression$group <- paired_phe$group
PRH2_expression$paired <- paired_phe$subject
PRH2_expression = PRH2_expression[order(as.numeric(paired_phe$subject)),] #这里需要按照配对信息排序好,以做后需的配对差异分析4.2 开始绘图
# 这个是我自己写的一个ggplot2的主题,可以自定义修改其中的参数
if(T){mytheme <- theme(plot.title = element_text(size = 12,color="black",hjust = 0.5),
axis.title = element_text(size = 12,color ="black"),
axis.text = element_text(size= 12,color = "black"),
panel.grid.minor.y = element_blank(),
panel.grid.minor.x = element_blank(),
panel.grid=element_blank(),
legend.position = "none",
legend.text = element_text(size= 12),
legend.title= element_text(size= 12)
)}
# 配对t检验箱式图图
box_paired <- ggplot(PRH2_expression, aes(x = group,y = PRH2,fill= group)) +
geom_boxplot(aes(fill = group),width= 1) +
geom_point(size= 1, alpha=0.6) +
geom_line(aes(group = paired), colour="black", linetype="11",size=0.5)+
scale_fill_manual(values = c("#1CB4B8", "#EB7369"))+
scale_y_continuous(limits=c(0, 25),breaks =seq(0,20,5),expand = c(0, 0) )+
labs(y="log2 Expression",x=NULL,title = "TCGA")+
theme_bw()+ mytheme +
stat_compare_means(
label = "p.format",
method = "t.test",
paired = T,
size = 4,
label.x = 1.5,
label.y = 22
)
box_paired
推荐一款高颜值免费在线SCI绘图工具~~~ 可以直接做配对箱线图
ggsave("./Output/TCGA_PRH2_paired_TumorvsNormal.PDF",
box_paired ,height = 8 ,width = 6,units = "cm")
sessionInfo()
## R version 4.0.2 (2020-06-22)
## Platform: x86_64-w64-mingw32/x64 (64-bit)
## Running under: Windows 10 x64 (build 14393)
##
## Matrix products: default
##
## locale:
## [1] LC_COLLATE=Chinese (Simplified)_China.936 LC_CTYPE=Chinese (Simplified)_China.936
## [3] LC_MONETARY=Chinese (Simplified)_China.936 LC_NUMERIC=C
## [5] LC_TIME=Chinese (Simplified)_China.936
##
## attached base packages:
## [1] parallel stats4 stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] ggthemes_4.2.0 ggpubr_0.4.0 ggplot2_3.3.2
## [4] dplyr_1.0.0 DESeq2_1.28.1 SummarizedExperiment_1.18.2
## [7] DelayedArray_0.14.0 matrixStats_0.56.0 Biobase_2.48.0
## [10] GenomicRanges_1.40.0 GenomeInfoDb_1.24.2 IRanges_2.22.2
## [13] S4Vectors_0.26.1 BiocGenerics_0.34.0
##
## loaded via a namespace (and not attached):
## [1] tidyr_1.1.0 bit64_0.9-7 splines_4.0.2 carData_3.0-4
## [5] blob_1.2.1 cellranger_1.1.0 GenomeInfoDbData_1.2.3 yaml_2.2.1
## [9] pillar_1.4.6 RSQLite_2.2.0 backports_1.1.7 lattice_0.20-41
## [13] glue_1.4.1 digest_0.6.25 RColorBrewer_1.1-2 ggsignif_0.6.0
## [17] XVector_0.28.0 colorspace_1.4-1 htmltools_0.5.0 Matrix_1.2-18
## [21] XML_3.99-0.4 pkgconfig_2.0.3 broom_0.7.0 haven_2.3.1
## [25] genefilter_1.70.0 zlibbioc_1.34.0 purrr_0.3.4 xtable_1.8-4
## [29] scales_1.1.1 openxlsx_4.1.5 rio_0.5.16 BiocParallel_1.22.0
## [33] tibble_3.0.2 annotate_1.66.0 farver_2.0.3 generics_0.1.0
## [37] car_3.0-8 ellipsis_0.3.1 withr_2.2.0 readxl_1.3.1
## [41] survival_3.1-12 magrittr_1.5 crayon_1.3.4 memoise_1.1.0
## [45] evaluate_0.14 rstatix_0.6.0 forcats_0.5.0 foreign_0.8-80
## [49] tools_4.0.2 data.table_1.12.8 hms_0.5.3 lifecycle_0.2.0
## [53] stringr_1.4.0 munsell_0.5.0 locfit_1.5-9.4 zip_2.1.1
## [57] AnnotationDbi_1.50.1 compiler_4.0.2 rlang_0.4.6 grid_4.0.2
## [61] RCurl_1.98-1.2 labeling_0.3 bitops_1.0-6 rmarkdown_2.3
## [65] gtable_0.3.0 abind_1.4-5 DBI_1.1.0 curl_4.3
## [69] R6_2.4.1 knitr_1.29 bit_1.1-15.2 stringi_1.4.6
## [73] Rcpp_1.0.5 vctrs_0.3.1 geneplotter_1.66.0 tidyselect_1.1.0
## [77] xfun_0.15参考资料
-
《差异分析|DESeq2完成配对样本的差异分析》
-
《R数据科学》
-
数据和代码下载:
https://gitee.com/ct5869/shengxin-baodian/tree/master/TCGA
-
作者:赵法明
编辑:生信宝典
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集



智能推荐
【BZOJ】3224: Tyvj 1728 普通平衡树-程序员宅基地
文章浏览阅读77次。【题意】1. 插入x数2. 删除x数(若有多个相同的数,因只删除一个)3. 查询x数的排名(若有多个相同的数,因输出最小的排名)4. 查询排名为x的数5. 求x的前驱(前驱定义为小于x,且最大的数)6. 求x的后继(后继定义为大于x,且最小的数)【算法】平衡树(treap)重要的细节以注释的形式标注在代码中。#include<cstdio>...
视图中的键保留表_视图键保留表-程序员宅基地
文章浏览阅读3.2k次。视图中的键保留表:连接视图中所有更新的列必须映射到键保留表的列中,也就是视图DML操作的列必须映射到键保留表的列中键保留表的理解是:一个复杂视图,若需要出现键保留表的话则必须保证基表中至少有一张表是有主键的! 其次,这两张表在进行关联时(可以是表连接也可以是多表查询,但一定要有关联条件,其关联条件其实相当于两表的主外键关系),如果关联条件是使用了主键的话,则外键表为键保留表_视图键保留表
java创建不定长数组_java二维不定长数组测试-程序员宅基地
文章浏览阅读209次。package foxe;import javax.swing.JEditorPane;import javax.swing.JFrame;/*** @author fooxe** @see:Test.java***/public class Test extends JFrame {private String arr[][] = null;private String str[][] = { ..._java创建一个不定长的数组
51信用卡Android 架构演进实践-程序员宅基地
文章浏览阅读227次。随着业务的快速扩张,原本小作坊式的单个工程的开发模式越来与不能满足实际需求。早在两年多以前,51信用卡管家就向下沉淀出了单独的公用基础库,一些通用的功能组件和个别独立的业务被拆分成 SDK,形成了一套中型项目、多人并行的开发模式,也为未来组件化拆分做准备。这套框架运行了一段时间之后,伴随着单应用内业务需求的增加、开发人员数量的增多、基础库数量的膨胀,导致了一些问题:主工程代码耦合严重,牵一发而动全...
机器学习模型评分总结(sklearn)_model.score-程序员宅基地
文章浏览阅读1.5w次,点赞10次,收藏129次。文章目录目录模型评估评价指标1.分类评价指标acc、recall、F1、混淆矩阵、分类综合报告1.准确率方式一:accuracy_score方式二:metrics2.召回率3.F1分数4.混淆矩阵5.分类报告6.kappa scoreROC1.ROC计算2.ROC曲线3.具体实例2.回归评价指标3.聚类评价指标1.Adjusted Rand index 调整兰德系数2.Mutual Informa..._model.score
Apache虚拟主机配置mod_jk_apache mod_jk 虚拟-程序员宅基地
文章浏览阅读344次。因工作需要,在Apache上使用,重新学习配置mod_jk1. 分别安装Apache和Tomcat:2. 编辑httpd-vhosts.conf: LoadModule jk_module modules/mod_jk.so #加载mod_jk模块 JkWorkersFile conf/workers.properties #添加worker信息 JkLogFil_apache mod_jk 虚拟
随便推点
小米组织架构再调整,王川调职,雷军自任中国区总裁_小米更换硬件负责人-程序员宅基地
文章浏览阅读335次。5月17日,小米集团再发组织架构调整及任命通知。新通知主要内容为前小米中国区负责人王川调职,雷军自任中国区总裁。小米频繁调整背后,雷军有些着急了中国区手机业务持续下滑。根据IDC最近公布的数据,小米一季度全球出货量为2750万台,相比去年同期的2780万台,小幅下降。参考Canalys、Counterpoint的统计,小米一季度出货量也都录得1%的同比下滑。作为对比,IDC数据显示,华为同期出..._小米更换硬件负责人
JAVA基础学习大全(笔记)_java学习笔记word-程序员宅基地
文章浏览阅读9.1w次。JAVASE和JAVAEE的区别JDK的安装路径[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-perPRPgq-1608641067105)(C:\Users\王东梁\AppData\Roaming\Typora\typora-user-images\image-20201222001641906.png)]卸载和安装JDK[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-SYnXvbAn-1608641067107)(C:\Users_java学习笔记word
vue-echarts饼图/柱状图点击事件_echarts 饼图点击事件-程序员宅基地
文章浏览阅读7.8k次,点赞2次,收藏17次。在实际的项目开发中,我们通常会用到Echarts来对数据进行展示,有时候需要用到Echarts的点击事件,增加系统的交互性,一般是点击Echarts图像的具体项来跳转路由并携带参数,当然也可以根据具体需求来做其他的业务逻辑。下面就Echarts图表的点击事件进行实现,文章省略了Echarts图的html代码,构建过程,option,适用的表格有饼图、柱状图、折线图。如果在实现过程中,遇到困难或者有说明好的建议,欢迎留言提问。_echarts 饼图点击事件
操作系统思维导图(一)_操作系统课程思维导图-程序员宅基地
文章浏览阅读1.3k次,点赞4次,收藏14次。内容整理自,华中科技大学,苏曙光老师《操作系统原理》,可在MOOC课程学习相关课程。_操作系统课程思维导图
vite build-程序员宅基地
文章浏览阅读4.3k次。vite在开发阶段采用的是按需加载的方式,不会将所有文件打包。但是生产环境的部署是需要进行打包的,这里它使用的是rollup打包方式。对于代码切割的需求,使用原生动态导入,因此打包后支持新浏览器,对IE的兼容性不是很好,但是可以用对应的polyfill解决。使用esbuild来处理需要pre-undle的在cli.ts的build命令中引入build.ts调用doBuild方法,在这个方法中配置打包参数(input output plugin等)调用buildHtmlPlugin解析文件入口in_vite build
Scala:访问修饰符、运算符和循环_scala ===运算符-程序员宅基地
文章浏览阅读1.4k次。http://blog.csdn.net/pipisorry/article/details/52902234Scala 访问修饰符Scala 访问修饰符基本和Java的一样,分别有:private,protected,public。如果没有指定访问修饰符符,默认情况下,Scala对象的访问级别都是 public。Scala 中的 private 限定符,比 Java 更严格,在嵌套类情况下,外层_scala ===运算符